










Join the Leading Global Eye Health Alliance.
Membership-
Choose an alternate language here
- Membership
 Los pacientes que sufren de enfermedades genéticas del ojo tienen necesidades especiales en términos de diagnóstico y manejo por ser entidades raras, necesidades para visión baja y consejo genético, así como necesidades para ajustes psicosociales. Las enfermedades raras genéticas afectan al menos a 1 en 50 individuos (http://orphanet.net) y el número total de esas entidades se ha estimado en 6000-7000, de las cuales cerca de un tercio pueden afectar al ojo (http://www.omim.org), lo cual significa que juntas esas entidades genéticas contribuyen significativamente en la morbi-mortalidad y en los costos de cualquier sistema de salud. Se ha estimado que alrededor de un 50% de los pacientes con estas enfermedades raras genéticas nunca reciben un diagnóstico (1) y se “embarcan“ en una situación denominada “odisea diagnóstica” que incluye múltiples consultas con especialistas, estudios de imagen, estudios de laboratorio e investigaciones invasivas (como biopsias). La odisea diagnóstica por definición es una “aventura” lenta, costosa, y también decepcionante porque simplemente no logra el propósito principal: el diagnóstico (2). Una encuesta de pacientes ha demostrado que el intervalo de diagnóstico de estos pacientes es entre 5-30 años, durante el cual hasta un 40% de los pacientes reciben diagnósticos incorrectos (3), con lo cual se incurre en un manejo, así como también procedimientos inadecuados (4). Aportar la confirmación diagnóstica molecularmente acorta dramáticamente el tiempo de la odisea diagnóstica, mejora el manejo, los tratamientos y la supervivencia de los pacientes. Del mismo modo, se mejoran los informes de un consejo genético con respecto a los riesgos de recurrencia y las opciones para una planificación familiar para los afectados y familiares. Nosotros describimos los beneficios de los estudios genéticos para pacientes con enfermedades hereditarias oftalmológicas.
Los pacientes que sufren de enfermedades genéticas del ojo tienen necesidades especiales en términos de diagnóstico y manejo por ser entidades raras, necesidades para visión baja y consejo genético, así como necesidades para ajustes psicosociales. Las enfermedades raras genéticas afectan al menos a 1 en 50 individuos (http://orphanet.net) y el número total de esas entidades se ha estimado en 6000-7000, de las cuales cerca de un tercio pueden afectar al ojo (http://www.omim.org), lo cual significa que juntas esas entidades genéticas contribuyen significativamente en la morbi-mortalidad y en los costos de cualquier sistema de salud. Se ha estimado que alrededor de un 50% de los pacientes con estas enfermedades raras genéticas nunca reciben un diagnóstico (1) y se “embarcan“ en una situación denominada “odisea diagnóstica” que incluye múltiples consultas con especialistas, estudios de imagen, estudios de laboratorio e investigaciones invasivas (como biopsias). La odisea diagnóstica por definición es una “aventura” lenta, costosa, y también decepcionante porque simplemente no logra el propósito principal: el diagnóstico (2). Una encuesta de pacientes ha demostrado que el intervalo de diagnóstico de estos pacientes es entre 5-30 años, durante el cual hasta un 40% de los pacientes reciben diagnósticos incorrectos (3), con lo cual se incurre en un manejo, así como también procedimientos inadecuados (4). Aportar la confirmación diagnóstica molecularmente acorta dramáticamente el tiempo de la odisea diagnóstica, mejora el manejo, los tratamientos y la supervivencia de los pacientes. Del mismo modo, se mejoran los informes de un consejo genético con respecto a los riesgos de recurrencia y las opciones para una planificación familiar para los afectados y familiares. Nosotros describimos los beneficios de los estudios genéticos para pacientes con enfermedades hereditarias oftalmológicas.
Beneficios de los estudios diagnósticos para los pacientes con enfermedades hereditarias del ojo
Caso 1. Familia costarricense originaria de un poblado lechero de la provincia de Alajuela. Padres consanguíneos, 8 hermanos de los cuales 4 tienen ceguera y 4 sanos. Los 4 afectados tienen entre 45 y 60 años de edad. Su problema de la visión inició desde su infancia; sin embargo, a pesar de su edad actual nunca les han comentado un diagnóstico y una causa de su ceguera, a pesar de que fueron multitratados desde la niñez. Esta familia fue estudiada genéticamente por medio de una tecnología denominada de microarreglos. Se identificó una variante patogénica en el gen GALK1 (galactocinasa 1), un gen relacionado a galactosemia que únicamente causa catarata. Esta enfermedad se puede prevenir y tratar sencillamente con el evitar la toma de leche y productos lácteos derivados. Los pacientes y familiares mostraron mucha satisfacción al conocer su diagnóstico. El estudio se extendió a toda la familia, se dio asesoramiento genético a la familia.
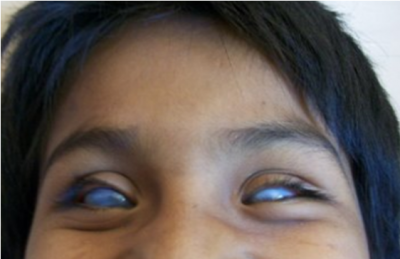
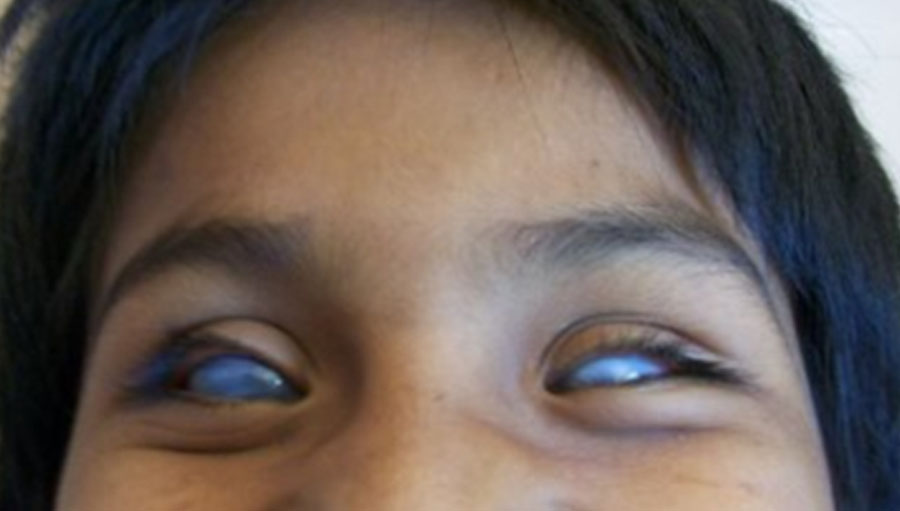
Caso 2. Una comunidad en el estado de Tlaxcala manifiesta por medios escritos (prensa estatal) su preocupación porque muchos de sus individuos manifiestan malformaciones en sus ojos, la cual concluye con ceguera. Se visitó la comunidad y se identificó a 22 afectados con una entidad clínica denominada esclerocórnea, afaquia y microftalmia (figura 1), dando una prevalencia de 2.52 casos por 1000 habitantes. La tasa de portadores fue de 1 en 40 habitantes de la comunidad. Se le ofrece a la comunidad un asesoramiento genético y medidas epidemiológicas para evitar que se repita la enfermedad.
Caso clínico 3. Las distrofias retinianas hereditarias son un grupo de enfermedades que afectan una célula o grupo de células retinianas específicas que afectan variablemente la visión. Su forma más severa se denomina amaurosis congénita de Leber (LCA) en la cual se han identificado más de 20 genes causantes de la enfermedad, dentro de ellos los genes RPE65, GUCY2D, y RDH12. La identificación del gen específico puede en muchos casos darnos un pronóstico visual. Así, pacientes con defectos en RPE65 tienen una mala visión en sus primeros años de vida, posteriormente exhiben una mejoría y permanece estable un tiempo, para posteriormente declinar. Por su parte, pacientes con LCA con defectos en RDH12 tienen un declive constante y progresivo de la visión. Por último, pacientes con mutaciones en GUCY2D exhiben una evolución estacionaria y visión estable durante mucho tiempo, antes de declinar.
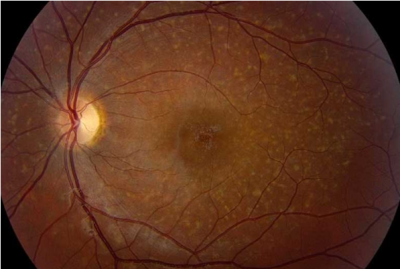
Caso 4. Masculino de 25 años de edad que inicia su padecimiento a los 8 años de edad con disminución de la agudeza visual. Posteriormente se agrega fotofobia y discromatopsia. En el examen de fondo de ojo se observan lesiones pisciformes o flecks en región macular. La fluorangiografía muestra “silencio coroideo” y la autofluorescencia atrofia macular (figura 2). Los datos identificados en este paciente nos dirigen al diagnóstico de la maculopatía hereditaria juvenil más frecuente, la enfermedad de Stargardt. El gen causal de estos pacientes se denomina ABCA4, y estudios en fase 2 ya se realizan en humanos para proveer una posible terapia.
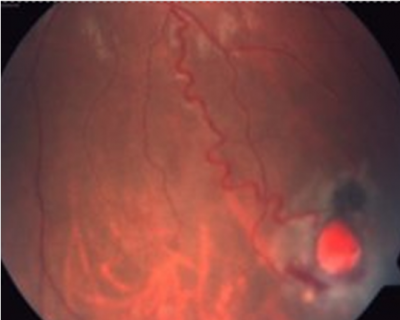
Caso 5. Femenino de 24 años de edad con diagnóstico de síndrome de von Hippel Lindau (entidad genética con patrón de herencia autosómica dominante que se caracteriza por tumoraciones en diversos sistemas de muestro cuerpo: hemangiomas retinianos, hemangioblastomas en cerebelo, feocromocitoma, carcinoma renal, tumores pancreáticos, entre otros) (figura 3) y un embarazo de 7 semanas de gestación. En esa misma fecha se le realizó un estudio genético molecular para secuenciar en gen VHL, a la siguiente semana se identificó una variante patogénica causante de la enfermedad. Se le realizó biopsia de vellosidades coriales para el estudio molecular prenatal. No se identificó la variante en el producto. Se confirma al nacer que el producto que no presenta la mutación. Se asistió a la joven pareja, el estudio demuestra un producto sin la enfermedad familiar.
Una de las principales razones para realizar el examen genético es preparar a los pacientes para en un futuro cercano la participación en ensayos clínicos para terapias que serán establecidas. En el año 2018, la FDA aprueba la primera terapia génica para pacientes con LCA con defectos en el gen RPE65. Actualmente hay más de 130 ensayos clínicos relacionados a distrofias retinianas, en la mayoría de ellos el conocimiento del gen causal para la enfermedad es de suma importancia para ser aceptado e incluido en este tipo de estudios.
La incorporación de los estudios genéticos puede contribuir favorablemente al cuidado de muchos pacientes con enfermedades hereditarias del ojo, aumentar las opciones terapéuticas y evaluar el riesgo de enfermedad para familiares. El examen genético-molecular debe ser considerado rutinariamente como parte de la evaluación clínica oftalmológica.
Oscar Francisco Chacón Camacho1,2
1 Unidad de Investigación, Departamento de Genética, Instituto de Oftalmología Conde de Valenciana
2 Departamento de Biología Molecular y Genética, Facultad de Estudios Superiores Iztacala, Universidad Autónoma de México
Correspondencia: Unidad de Investigación, Departamento de Genética, Instituto de Oftalmología Conde de Valenciana. Chimalpopoca N° 14, Colonia Obrera, Delegación Cuauhtemoc, Ciudad de México, México. CP 06800. Email: oscar_chacon73@hotmail.com. Tel: 5554421700 ext 3212.









